



Соединения четырехкоординированного фосфора состава Y = P(NR2)3 представляют большой интерес в качестве лигандов в координационной химии переходных металлов и в качестве молекулярных систем для разработки лекарственных препаратов, корректирующих метаболические процессы в организме человека [5, 9]. При интерпретации данных фотоэлектронной (ФЭ) спектроскопии фосфорсодержащих соединений [6, 7] возникают проблемы с отнесением некоторых спектральных полос, связанных с тонкими эффектами и не подлежащих объяснению в рамках стандартных подходов. Одним из возможных эффектов, проявляющихся в спектрах, является широко изучаемый и применяемый на практике эффект образования ассоциатов молекулярных систем [3, 8, 10, 11].
Целью настоящей работы является квантово-химическое исследование влияния димеризации на электронное строение и ФЭ спектр гексаметилтиофосфортриамида SP(NMe2)3.
Материалы и методы исследования
Квантово-химические расчеты выполнены с помощью программы GAMESS-US (версия 05.12.2014) [13] в вакуумном приближении методами Хартри – Фока HF/6-311++G** [2, 4] и теории функционала плотности (ТФП) DFT/РBЕ0/6-311++G** [1]. Геометрические параметры полностью оптимизировались для мономера, димера и тетрамера молекулы в основном состоянии (рис. 1), условие минимума энергии определялось по гессиану. Сделана оценка суперпозиционной ошибки базисного набора при расчете энергии образования димера и тетрамера (SP(NMe2)3)n, несмотря на то, что этот вид ошибки является нефизическим эффектом и вопрос о её учете остается дискуссионным [12]. Согласно проведенным расчетам, величина данной ошибки не превышает 5 %. Результаты расчетов визуализировались с помощью программы Chemcraft 1.8 (b404) [15].
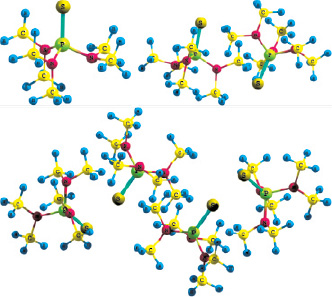
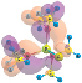
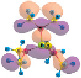
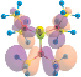
Рис. 1. Рассчитанные молекулярные системы (SP(NMe2)3)n (n = 1, 2, 4), HF/6-311++G**
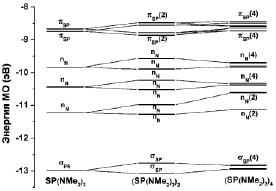
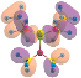
Рис. 2. Корреляционная диаграмма энергий МО при образовании димеров и тетрамеров соединения SP(NMe2)3, HF/6-311++G**.
Результаты исследования и их обсуждение
Анализ полученных данных показал, что метод HF/6-311++G** корректнее, чем DFT/РBE0/6-311++G**, воспроизводит энергии верхних занятых молекулярных орбиталей (ВЗМО), ассоциируемых с вертикальными потенциалами ионизации соединения (табл. 1), что объясняется нарушением теоремы Купманса в приближении ТФП. Тем не менее метод DFT/RBE0/6-311++G** качественно верно описывает основные тенденции изменения состава и энергий МО при образовании димеров и тетрамеров молекулы S = P(NMe2)3. Согласно проведенным расчетам молекулы в экспериментальной и оптимальной геометрии, образование димера (SP(NMe2)3)2 и тетрамера (SP(NMe2)3)4 приводит к изменениям энергий и состава ВЗМО, включая МО с доминирующим вкладом валентных АО азота (рис. 2, табл. 1, 2). При этом стабилизации МО неподеленных электронных пар (НЭП) азота не происходит, а напротив, наблюдается дестабилизация электронов на πSP-, nN- и σPS-МО. Очевидно, что изменения энергии верхних занятых МО, включая МО неподеленных электронных пар азота, будут связаны с уширением соответствующих спектральных полос.
Таблица 1
Энергии связи Ii и рассчитанные энергии -εi (эВ) ВЗMO соединения SP(NMe2)3
|
№ MO |
Вид MO a |
Тип MO a |
Iib |
–εic |
–εia |
–εid |
–εie |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
53 |
|
πSP (69 %), nN (18 %) |
7,66 (3pπS) |
8,41 |
8,67 |
5,80 |
6,18 |
|
52 |
|
πSP (47 %), nN (39 %) |
8,05 (3pπS) |
8,52 |
8,75 |
5,93 |
6,22 |
|
51 |
|
nN (70 %) |
8,28 (2pN) |
9,98 |
9,83 |
6,69 |
6,69 |
|
50 |
|
nN (53 %), σSP (23 %) |
8,64 (2pN) |
10,47 |
10,44 |
7,05 |
7,09 |
|
49 |
|
nN (51 %) σPS (32 %) |
9,14 (2pN) |
10,84 |
11,23 |
7,45 |
7,85 |
|
48 |
|
σPS (84 %) |
9,61 (2pN) |
12,88 |
12,98 |
9,64 |
9,86 |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
47 |
|
σPN (55 %), σCH (39 %) |
11,72 (C-H, P-N) |
14,39 |
14,18 |
10,74 |
10,45 |
|
46 |
|
σCH (63 %), σNC (29 %) |
12,40 (C-H, P-N) |
14,62 |
14,20 |
10,91 |
10,47 |
|
45 |
|
σCH (78 %), σNC (17 %) |
13,70 (C-H, P-N) |
14,91 |
14,25 |
11,06 |
10,48 |
|
44 |
|
σCH (61 %), σPN (39 %) |
14,80 (C-H, P-N) |
15,13 |
14,47 |
11,23 |
10,68 |




Примечания:
a HF, оптимальная геометрия; b данные ФЭС [14]; c HF, экспериментальная геометрия;
d DFT/РBЕ0, экспериментальная геометрия; e DFT/РBЕ0, оптимальная геометрия; базисный набор 6-311++G**.
Таблица 2
Рассчитанные энергии -εi а (эВ) и тип ВЗMO молекулярных систем (SP(NMe2)3)n
|
(S = P(NMe2)3)2 |
(S = P(NMe2)3)4 |
||
|
-εi |
Тип МО |
-εi |
Тип МО |
|
1 |
2 |
3 |
4 |
|
8,48; 8,56 |
πSP (43 %), σCH (41 %); πSP (47 %), nN (28 %) |
8,45; 8,49; 8,58; 8,59 |
πSP, nN; πSP, nN, σCH; πPS, σCH, nN; πPS, σCH, nN; |
|
8,78; 8,86 |
πSP (51 %), σCH (26 %), nN (15 %); πSP (44 %), nN (31 %), σCH (16 %) |
8,65; 8,65; 8,73; 8,73 |
πSP, σCH, nN; πSP, σCH, nN; πSP, nN, σCH; πSP, nN, σCH |
|
9,58; 9,89 |
nN (57 %), σCH (26 %); nN (51 %), σCH (23 %), πPS (18 %) |
9,69; 9,72; 9,80; 9,83 |
nN, πPS, σCH; nN, πPS, σCH; nN, σCH, πPS; nN, πPS, σCH |
|
10,23; 10,52 |
nN (46 %), σPS (24 %), σCH (16 %); nN (41 %), σCH (30 %), σPS (21 %) |
10,33; 10,33; 10,37; 10,38 |
nN, σCH, σPS; nN, σCH, σPS; nN, σCH, σSP; nN, σCH, σSP |
|
10,98; 11,27 |
nN (42 %), σCH (36 %), σPS (15 %); nN (54 %), σCH (24 %), σPS (19 %) |
10,60; 10,62; 11,13; 11,13 |
nN, σCH, σPS; nN, σPS, σCH; nN, σPS, σCH; nN, σPS, σCH; |
|
12,76; 13,08 |
σSP (70 %), nN (12 %), σCH (12 %); σSP (66 %), σCH (16 %), nN (12 %) |
12,82; 12,83; 12,93; 12,94 |
σSP, nN, σCH; σSP, σCH, nN; σSP, nN, σCH; σSP, nN, σCH |
|
13,96; 13,96 |
σCH (65 %), σPN (33 %); σCH (54 %), σPN (39 %) |
14,00; 14,03; 14,03; 14,04 |
σCH, σPN; σPN, σCH; σPN, σCH; σCH, σPN |
|
14,01; 14,21 |
σCH (60 %), σPN (36 %); σCH (48 %), σPN (43 %) |
14,05; 14,05; 14,09; 14,10 |
σCH, σPN; σCH, σPN; σCH, σPN; σCH, σPN |
|
14,23; 14,28 |
σCH (50 %), σPN (39 %); σCH (72 %), σPN (22 %) |
14,13; 14,13; 14,15; 14,15 |
σCH, σPN; σCH, σPN; σCH, σPN; σCH, σPN |
|
14,29; 14,31 |
σCH (57 %), σPN (33 %); σNC (54 %), σCH (21 %), σPS (11 %) |
14,22; 14,23; 14,34; 14,35 |
σCH, σPN; σCH, σPN; σNC, σCH; σNC, σCH |
|
14,52; 14,58 |
σCH (61 %), σPN (36 %); σNC (57 %), σCH (21 %), σPS (14 %) |
14,37; 14,38; 14,45; 14,45 |
σCH, σPN; σCH, σPN; σNC, σCH, σPS; σNC, σCH, σPS |
|
14,70; 14,91 |
σCH (76 %); σCH (89 %) |
14,61; 14,74; 14,81; 14,81 |
σCH; σCH; σCH; σCH |
|
14,97; 15,02 |
σCH (83 %); σCH (85 %) |
14,93; 14,94; 15,04; 15,05 |
σCH; σCH; σCH; σCH |
Примечание. а HF/6-311++G**, оптимальная геометрия.
Установлено, что наблюдаемые изменения энергии и состава МО при образовании димеров и тетрамеров соединения вызваны значительным перераспределением электронной плотности, изменением порядков связей и уменьшением полярности молекулярной системы (дипольный момент мономера, димера и тетрамера равен 5,2; 4,5 и 0,1 Д соответственно, HF/6-311++G**).
Исследование влияния температурных эффектов на электронное строение и ФЭ спектры гексаметилтиофосфортриамида может быть проведено с помощью методов молекулярной динамики наряду с более корректным учетом корреляционных и дисперсионных эффектов для данного перспективного соединения.
Выводы
Квантово-химическое моделирование методами HF/6-311++G** и DFT/РBE0/6-311++G** в вакуумном приближении показало, что процессы образования молекулярных ассоциатов гексаметилтиофосфортриамида в газовой фазе существенно влияют на его электронное строение и, вероятно, на его ФЭ спектр. Можно предположить, что изменения энергии ВЗМО, включая МО НЭП азота, будут связаны с уширением соответствующих спектральных полос. Установлено, что наблюдаемые изменения энергии и состава МО вызваны значительным перераспределением электронной плотности молекулы.
Работа проводилась при финансовой поддержке Министерства образования и науки Российской федерации в рамках государственного контракта № 2014/36 с Дальневосточным федеральным университетом (проект № 1137).
Рецензенты:
Кавун В.Я., д.х.н., заведующий лабораторией химической радиоспектроскопии, ФГБУН «Институт химии» ДВО РАН, г. Владивосток;
Мирочник А.Г., д.х.н., заведующий лабораторией светотрансформирующих материалов, ФГБУН «Институт химии» ДВО РАН, г. Владивосток.