



Перспективность ряда хелатных комплексов бора, β-дикетонатов и β-кетоеминатов дифторида бора в плане практического применения в устройствах нелинейной оптики определяется их интенсивной люминесценцией [1–3, 10, 11]. Возбужденные состояния соединений этих классов связаны с образованием разнообразных короткоживущих переходных состояний [4, 6]. Значительный научный интерес представляют теоретические данные о самоорганизации этих систем, а именно о возникновении молекулярных ассоциатов в жидкой и твердой фазах. Большую практическую значимость имеет учет этих эффектов при описании возбужденных состояний хелатных комплексов дифторида бора, что определяет ценность квантово-химического моделирования возбуждения колебательно-вращательных и электронных переходов на начальных этапах физико-химических исследований данных молекулярных систем.
Ранее авторами проведены квантово-химическое моделирование электронной структуры, возбужденных состояний и спектров поглощения, а также экспериментальное исследование электронных переходов в некоторых хелатах дифторида бора [4] и комплексах иттрия, лантана (III) состава М(NO3)3(ГМФА)3 [5–8].
Целью настоящей работы является теоретическое физико-химическое исследование структуры, электронного строения, возбужденных состояний и спектрального поведения следующих хелатных комплексов дифторида бора: 2,2-дифторо-4-метилфенил-6-метил-1,3,2-диоксаборин (толуилацетонат дифторида бора), (p-CH3C6H4COCHCOCH3)BF2 (I) и 2,2-дифторо-4-метилоксифенил-6-метил-1,3,2-диоксаборин (анизоилацетонат дифторида бора), (p-CH3ОC6H4COCHCOCH3)BF2 (II).
Материалы и методы исследования
С помощью программы GAMESS-US [12] в вакуумном приближении в базисе 6-311G** неэмпирическим методом и методами функционала плотности DFT и TDDFT с гибридным обменно-корреляционным функционалом B3LYP [9] выполнены квантово-химические расчеты электронного строения и физико-химических свойств хелатных комплексов I и II в основном и возбужденных состояниях. Выбор функционала B3LYP связан с хорошим воспроизведением электронного строения соединений элементов I–III периодов. Для энергетически наиболее выгодных конфигураций была учтена релаксация молекулярной системы при переходе в возбужденное синглетное состояние.
Результаты исследования и их обсуждение
Согласно квантово-химическим расчетам неэмпирическим методом и методом DFT, молекулярные системы I, II являются сильно полярными, их дипольный момент в оптимальной геометрии равен 7,8 Д. Для изучения возбужденных состояний комплексов проведен анализ состава граничных молекулярных орбиталей (МО) (рис. 1).
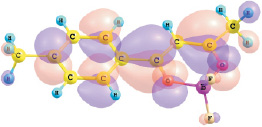 εНВМО, I = –2.62 эВ
εНВМО, I = –2.62 эВ
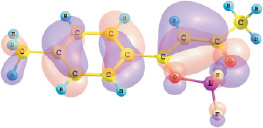 εВЗМО, I = –6.96 эВ
εВЗМО, I = –6.96 эВ
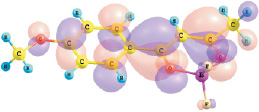 εНВМО, II = –2.44 эВ
εНВМО, II = –2.44 эВ
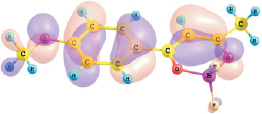 εВЗМО, II = –6.54 эВ
εВЗМО, II = –6.54 эВ
Рис. 1. Энергии и вид граничных МО комплексов I и II (DFT/B3LYP/6-311G**)
Установлено, что верхняя занятая МО (ВЗМО) комплекса I с энергией ε = –6,96 эВ и комплекса II с энергией –6,54 эВ является π-МО, в основном характеризующей π–системы хелатного и ароматического колец. Нижняя вакантная МО (НВМО) комплекса I с энергией –2,62 эВ и комплекса II с энергией –2,44 эВ является π*-МО, также в основном характеризующей π–системы хелатного и ароматического колец. При переходе от толуила к анизоилу энергетическая щель ВЗМО–НВМО рассматриваемых комплексов дифторида бора уменьшается от 4,34 до 4,10 эВ.
Методом TDDFT/B3LYP/6-311G** проведены квантово-химические расчеты 20 возбужденных синглетных и триплетных состояний хелатных комплексов I и II, смоделированы их УФ-спектры поглощения (рис. 2, 3). Анализ электронных синглет–синглетных переходов данных комплексов показал, что синглетное возбуждение молекулы I в области λпогл = 307 нм и молекулы II в области λпогл = 321 нм на 99 % обусловлено переходами валентных электронов с ВЗМО, соответствующей π–системам хелатного и ароматического колец, на вакантные молекулярные уровни, соответствующие НВМО-несвязывающей π*-МО π–систем хелатного и ароматического колец.
Учет релаксации молекулярных систем в возбужденном синглетном состоянии дает снижение энергии возбужденного состояния для молекулы I на 0,45 эВ и, соответственно, увеличение длины волны максимума электронного спектра на 38 нм, для молекулы II на 0,14 эВ и увеличение длины волны максимума электронного спектра на 12 нм.
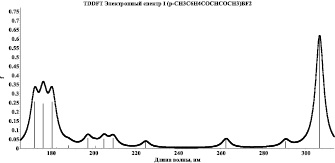
Рис. 2. Модельный спектр поглощения комплекса I
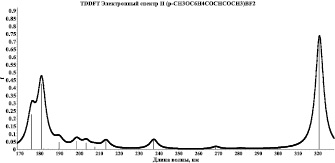
Рис. 3. Модельный спектр поглощения комплекса II
Таким образом, наиболее вероятные центры электронного возбуждения при переходе комплексов I и II в возбужденные состояния локализованы на π–системах хелатного и ароматического колец. На основании этого можно предположить, что широкая полоса с максимальной интенсивностью экспериментальных спектров поглощения данных комплексов обусловлена электронными переходами в указанных π–системах.
Выводы
Неэмпирическим методом и методами функционала плотности DFT и TDDFT с гибридным обменно-корреляционным функционалом B3LYP [9] в базисе 6-311G** выполнены квантово-химические расчеты электронного строения и физико-химических свойств комплексов дифторида бора I, II в основном и возбужденных состояниях. Для энергетически наиболее выгодных конфигураций была учтена релаксация молекулярной системы при переходе в возбужденное синглетное состояние. Показано, что изученные молекулярные системы являются полярными, их дипольный момент в оптимальной геометрии приблизительно равен 7,8 Д. На основе квантово-химического моделирования дана интерпретация особенностей возбужденных электронных состояний хелатных комплексов I, II.
Работа проводилась при частичной финансовой поддержке Министерства образования и науки Российской Федерации в рамках государственного задания Дальневосточного федерального университета № 3.8646.2013.
Рецензенты:
Гончарук В.К., д.х.н., профессор, заведующий лабораторией оптических материалов, ФГБУН «Институт химии ДВО РАН», г. Владивосток;
Кавун В.Я., д.х.н., заведующий лабораторией химической радиоспектроскопии, ФГБУН «Институт химии ДВО РАН», г. Владивосток.
Работа поступила в редакцию 19.12.2013.